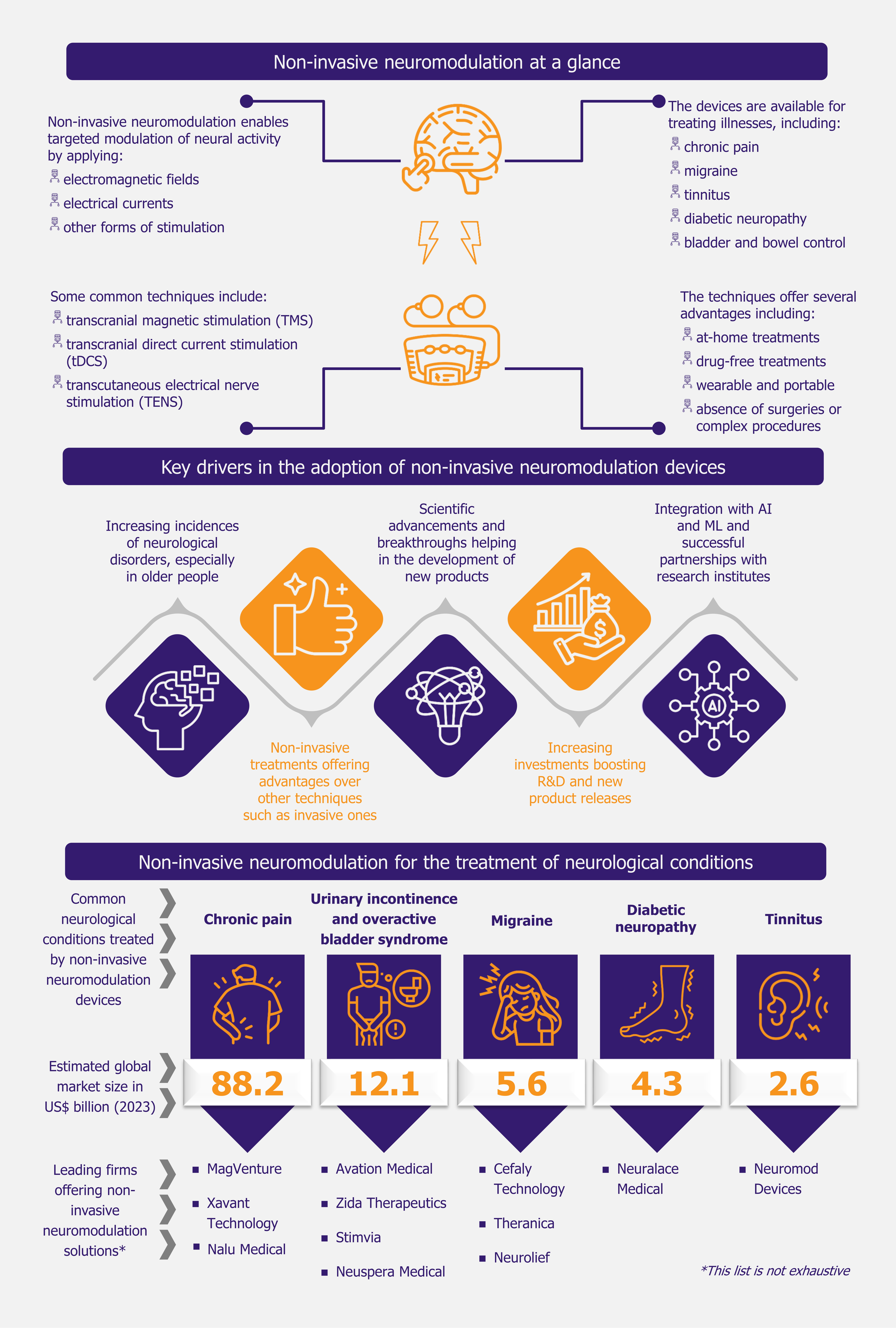



For many years, bariatric surgery has been the go-to option for people struggling with obesity and obesity-induced conditions. However, for the last couple of years, another easier option has become available in the form of GLP-1-based weight loss drugs. This class of medicine mimics a hormone that helps reduce food intake and control appetite. These drugs have revolutionized the weight loss market, which was previously dominated by gimmicky and fad-based OTC solutions. Due to GLP-1’s proven effectiveness, there is soaring demand for these drugs, outstripping its current supply capacity. While only two players operate in this market, several leading drugmakers have been racing to develop their own version of the drug. Moreover, with additional proven merits of the drug beyond just weight loss, it has become more appealing for pharma players to invest in.
GLP-1 anti-obesity drugs make big waves in the pharmaceutical sector
Over the past few years, anti-obesity drugs have received immense attention from healthcare professionals, pharmaceutical companies, and the general public. A new class of medication that stands out is glucagon-like peptide-1 (GLP-1) agonists, traditionally used for treating Type 2 diabetes. But along with managing diabetes, these drugs also suppress appetite and lower calorie intake by mimicking the GLP-1 hormone (a gastrointestinal hormone), which causes the patient to feel fuller longer and thereby prevents overconsumption. Regular intake of such drugs is deemed to result in a weight loss of about 15-25% of body weight in obese people.
GLP-1 agonists received FDA approval as anti-obesity drugs in 2021. Given their promising results, the demand for these drugs has increased immensely. However, despite the patient’s high out-of-pocket price of US$1,000 plus, there are severe shortages in the market.

Anti-Obesity Drugs – Pharma Companies Race to Grab a Bite of the Pie by EOS Intelligence
Only two players operate in this highly-coveted market
The GLP-1-based medication is now marketed in two categories – one for managing diabetes and blood sugar levels and the other as a weight loss drug. The GLP-1-based weight loss drug market is highly consolidated, as only two players operate in this space. These are Denmark-based Novo Nordisk and US-based Eli Lilly.
Novo Nordisk, the market leader, received FDA approval for its weight loss injectable, Wegovy, in June 2021. This drug uses the same active ingredient as Novo Nordisk’s diabetes drugs, Ozempic and Rybelsus (oral); however, it has a different dosage and can also be used for weight loss in patients who do not have diabetes. That being said, Ozempic has also been used off-label for weight loss.
On the other hand, Eli Lilly’s injectable drug for weight loss, Zepbound, received FDA approval in November 2023. Eli Lilly’s glucose-dependent insulinotropic polypeptide – GIP/GLP-1 injectable drug for diabetes, Mounjaro, has the same composition and dosage as Zepbound and is often prescribed off-label for weight loss as well.
While Novo Nordisk’s drugs, which use semaglutide as an active ingredient, result in weight loss of about 13 to 22 lbs, the drugs by Eli Lilly have tirzepatide as an active ingredient. They are stated to result in a weight loss ranging between 15 and 28 lbs.
From a price-point perspective, Wegovy has an out-of-pocket cost of US$1,349 per month, compared to Zepbound, which has an out-of-pocket cost of US$1,060 per month. Thus, while Novo Nordisk’s Wegovy has the first-mover advantage, Eli Lilly’s Zepbound is considered more effective and better priced.
Currently, both weight loss drugs by Novo Nordisk and Eli Lilly come in the form of injectables. However, both companies are developing oral versions of the drug as they are easier to administer and more convenient to prescribe. They may also help ease supply constraints currently impacting the injectables. In June 2023, Novo Nordisk conducted Phase 3 trials for its once-daily oral Wegovy drug, according to which the drug helped obese adults lose about 15% of their body weight. Similarly, in June 2023, Eli Lilly conducted Phase 2 trials for its oral GLP-1 receptor for weight loss. The drug helped obese adults lose up to 14.7% of their body weight. Both companies are optimistic about the outcomes of their trials; however, the expected launch timelines for these drugs have yet to be determined.
Leading drugmakers race to compete in the growing anti-obesity drug market
Currently, Novo Nordisk and Eli Lilly are the only two players operating in this market. However, several other leading pharmaceutical players have joined the race and are working towards developing their own version of the drug, either through in-house R&D or through strategic acquisitions.
Moreover, they are targeting their research towards developing and marketing a new generation of GLP-1-based medications that are administered orally, are longer lasting, and have additional health benefits and limited side effects.
In February 2024, US-based biopharmaceutical company Amgen successfully completed a Phase 1 clinical trial for its GLP-1 agonist drug, MariTide. As per the trials, the drug produced a 14.5% weight loss in patients administered the highest dose. Moreover, the company claims that the trial indicates that patients may need to take less frequent doses of MariTide (compared with current competition), and the weight loss achieved stays significantly longer. The company has begun its Phase 2 trial, with results expected by late 2024.
In December 2023, Swiss-pharmaceutical giant Roche acquired US-based Carmot for US$3.1 billion (US$2.7 billion upfront cash and US$400 million on certain milestones). This acquisition has helped put Roche on the map for obesity drug development. Carmot has two GLP-1 agonist molecules for weight loss, which are currently being tested in the mid to advanced stages of clinical trials. The first drug, CT-388, is a once-weekly injectable and has completed Phase 1 clinical trial, while the other drug, CT-996, is an oral drug currently undergoing Phase 1 trials.
In November 2023, UK drugmaker AstraZeneca entered into an agreement with Shanghai-based Eccogene, wherein the former licensed an oral once-daily GLP-1 receptor agonist called ECC5004 for the treatment of obesity, Type 2 diabetes, and other cardiometabolic conditions. For this, AstraZeneca agreed to pay Eccogene an upfront fee of US$185 million for the drug and a further payment of US$1.83 billion in future clinical, regulatory, and commercial milestones and tiered royalties. The drug is currently in Phase 1 development, and the company hopes to enter Phase 2 of clinical studies by the end of 2024. In the past, AstraZeneca stopped the development of two GLP-1 agonist drugs that were being developed in-house. The development of an injectable called Cotadutide was halted in April 2023, and an oral drug called AZD0186 was halted in June 2023 after their respective Phase 2b and Phase 1 clinical trials did not yield the desired results.
Pfizer, one of the most active companies in this regard, has faced multiple failures in their endeavor to develop a competitive obesity drug. In 2020, it started a clinical trial for its GLP-1 agonist weight loss drug, Lotiglipron. However, in June 2023, the company stopped developing the drug after its Phase 1 and Phase 2 drug interaction studies indicated a rise in liver enzymes in patients who took the drug once a day. In 2021, the company simultaneously began working on another GLP-1 receptor agonist, Danuglipron, which was to be taken twice daily. While the Phase 2a trial for the drug in June 2023 showed promise, the company halted the development of the drug post its Phase 2b trial in December 2023. The drug was scrapped as, despite significant weight loss, the trial patients experienced high rates of common gastrointestinal and mechanism-based adverse side effects. The company is now conducting a pharmacokinetic study with a once-daily version of the Danuglipron drug that will provide guidance on future development plans.
Pfizer’s failure with these two drugs demonstrates the struggle the leading pharma companies face to develop a safe, effective, and tolerable GLP-1 agonist for weight loss.
GLP-1 agonist drugs have benefits beyond diabetes and weight loss
Despite multiple setbacks, leading pharma companies are investing heavily in this space, as they understand the potential of these drugs. While currently, GLP-1 agonists are poised as diabetes and weight loss drugs, they have far more benefits. Data from ongoing clinical trials and independent studies suggest that GLP-1 agonists also help improve cardiovascular health and kidney function and help treat addiction and dementia.
In March 2024, Novo Nordisk’s Wegovy received FDA approval for reducing the risk of serious cardiovascular complications in adults with obesity and heart disease. This is based on the results shared from the company’s three-phase trial SELECT, which indicated that Wegovy reduced patients’ risk of major cardiovascular problems by about 20% during the five-year trial period.
Similarly, in 2019, the company started another clinical trial, FLOW, to determine the impact of GLP-1 agonists on kidney function. As per the interim results in October 2023, the trial displayed that Ozempic (Wegovy’s diabetes counterpart) reduced the risk of kidney disease progression and kidney and cardiovascular death in diabetes patients by 24%. Given its success, the company has halted the trial at the interim stage.
An initial study conducted on animals in March 2023 reportedly showed positive results for curbing addictive tendencies, such as drinking and smoking, with Ozempic. Currently, two trials are being undertaken to validate the use of GLP-1 agonists in humans to manage drug and alcohol addiction. Given the testimonies from current users of the drug, it is indicative that the drug has been helping users curb their addictions.
In addition to this, several researchers are also suggesting that GLP-1 could be used in the treatment of dementia and other cognitive disorders. This is based on the claim that GLP-1 agonists reduce the build-up of two proteins, amyloid, and tau, in the brain. These two proteins are known to be responsible for Alzheimer’s disease, which is the most common form of dementia. In February 2022, a new trial at the University of Oxford was initiated to test people with high levels of amyloid and tau and at risk of developing dementia to determine if the use of GLP-1 agonists would result in a reduction in tau accumulation and brain inflammation. The interim results from the study have not yet been disclosed.
High prices and limited coverage pose as speedbumps for obesity drug adoption
While these obesity drugs have exploded in popularity in recent times and are only expected to grow further as their case use increases, they do have certain shortcomings and challenges that are important to address.
These drugs are known to cause several side effects, such as nausea, diarrhea, vomiting, constipation, and ulcers. They can also lead to severe complications, such as pancreatitis, in some extreme cases. While most of the common side effects of the drugs are manageable and justifiable given the risk-benefit ratio, one of the key issues with the drugs is that they need to be taken in perpetuity to keep the weight off. In other words, once a patient stops taking the drugs, the weight comes back. Given that these drugs are priced at more than US$1,000 per month at the moment, taking them constantly becomes a considerable challenge for patients.
Moreover, considered as ‘vanity-use’, these drugs are currently not covered by most medical insurance policies, and thus, patients have to pay for them out-of-pocket. While several employers in the USA are considering including these drugs in their health plans, they are still debating their merit. Employers acknowledge the benefits of these drugs as they help employees who battle with obesity improve their health and, in turn, improve overall performance and employee satisfaction. However, high costs and long-term use act as definite barriers, which make both employers and insurers reluctant to cover these drugs.
Insurers are slowly warming up to the inclusion of GLP-1 drugs in their plans
In March 2024, leading insurance company Cigna stated that it would expand insurance coverage to include weight loss drugs but would limit how much health plans and employers spend on the drug each year. As per Cigna’s benefits management unit, Evernorth Health Services, spending increases for these weight loss and diabetes drugs would be limited to a maximum of 15% annually. The plan offers a financial guarantee and enables employers and health plans to have greater predictability and control over their GLP-1 spending by offering clients (employers) a guarantee that the cost of weight loss and diabetes drugs would not increase by more than 15% annually.
As a part of the effort to limit how much employers spend on GLP-1-based drugs annually, Evernorth has entered into an agreement with Novo Nordisk and Eli Lilly. However, the details of the agreement have not been disclosed.
While this is a good start, the drug would need better coverage by many other insurance players to reach a wider audience.
EOS Perspective
Given that about 12% of the global population and more than 40% of the American population grapple with obesity (as per WHO and 2022 statistics by the National Institute of Diabetes and Digestive and Kidney Diseases, USA, respectively), weight loss drug manufacturers Novo Nordisk and Eli Lilly are sitting on pharma goldmines. The weight loss drugs market, expected to reach US$100 billion by 2030, is poised as one of the most promising sectors for the pharma sector. Thus, it is no surprise that several leading players are investing heavily to join Novo Nordisk and Eli Lilly at the top, either through in-house R&D or through acquisitions.
However, developing these drugs proves to be challenging for drugmakers, as evidenced by the failures of several companies in creating their own versions. We can expect the sector to consolidate further as larger pharma companies look to acquire niche players with their trials being in advanced stages.
Moreover, in a bid to find their footing in this promising sector, pharma players are trying to develop advanced versions of the drug that have benefits beyond just weight loss and offer long-term benefits. This is also because, at the moment, these drugs are not approved by most insurance companies, which makes them extremely expensive for the wider population to afford. This, in turn, is withholding these drugs from becoming mainstream and is thereby preventing them from tapping into their true growth potential. That being said, Wegovy’s recent FDA approval for reducing cardiac complications in people with obesity and heart disease will likely tip the insurers’ coverage scales. Insurance companies are likely to cover the drug in the near future.
Since no other drug in the market offers proven cardiac benefits along with weight loss (including Eli Lilly’s), it is safe to say that Novo Nordisk is way ahead in the race and will dominate the market for the foreseeable future. Thus, to be able to compete in the market, it is not enough for drugmakers to develop obesity drugs offering just weight benefits. They would need to develop drugs that offer higher efficiency or additional therapeutic benefits along with weight loss and price them competitively.