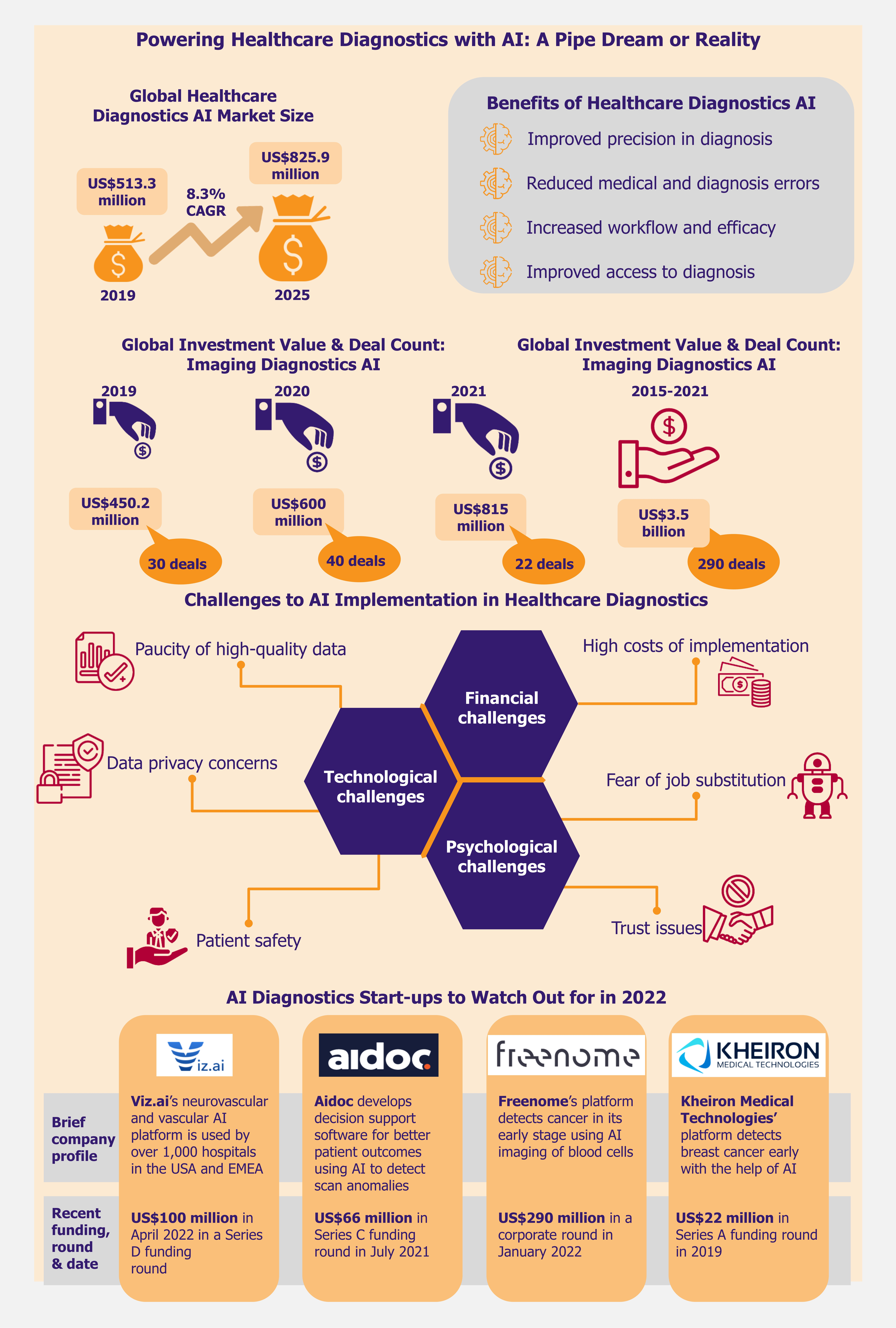
Pulsed Field Ablation (PFA) is an emerging technology for treating atrial fibrillation (AFib), a form of irregular heartbeat affecting 40 million heart patients worldwide as of 2023. As the prevalence of AFib is increasing, all eyes are on this novel, minimally invasive technology that offers improved effectiveness, safety, and shorter procedure and recovery time compared to the existing thermal ablation procedures.
PFA applies short, high-voltage pulses of energy to cardiac tissue and is proven to be more precise and safe than the thermal ablation methods, which come with the risk of damaging collateral tissues.
A clinical trial conducted by Medtronic across North America, Europe, Australia, and Japan during 2022-2023 revealed that the efficacy performance of its PFA system PulseSelect stood at 66% in paroxysmal and 55% in persistent AFib patients against the pre-specified performance goals of >50% (paroxysmal) and >40% (persistent). Performance goals were set based on multiple studies conducted on thermal ablation procedures that evaluated efficacy based on the freedom from acute procedural failure and arrhythmia recurrence in one year.
Despite promising results, the first-generation PFA technology still needs improvement in targeting the tissue of interest, and players in the field are developing supportive systems such as mapping systems to improve performance.
PFA emerges as a better alternative to conventional ablation methods
PFA is viewed as the best evolution within the electrophysiology (EP) space (comprises ablation catheters, diagnostic catheters, laboratory devices, and access systems used to treat arrhythmia). The tissue-targeting approach of PFA overcomes the drawbacks of thermal ablation methods, such as extensive scarring and the risks of injuring nearby organs. Along with improving clinical outcomes, this transformative technology will significantly improve patient experience and reduce the cost of care by lowering procedure and recovery time.
Being safer than other ablation methods, PFA is set to become the preferred modality
Only about 2% of the eligible patients with AFib globally and 15% of the eligible patients in the USA were treated with ablation as of February 2023, according to a MedTech analyst at Bank of America. This is because thermal ablation comes with the risk of damaging nearby issues, which can lead to damage to the esophagus, phrenic nerve, and pulmonary veins.
A study published by the European Heart Rhythm Association in January 2024 comparing the outcomes of PFA and thermal ablations stated that the risk of injury from PFA was 3.4% compared to 5.5% in thermal ablation. PFA, being safer than thermal ablation, can be expected to reach many more eligible patients. After the launch of Boston Scientific’s Farapulse in Europe in January 2021, 38,000 AFib patients were treated there with the Farapulse PFA system during 2022-2023, compared to 2,000 patients Farapulse treated in 2021. Moreover, Boston Scientific predicts the global AFib ablation market will grow from US$5 billion in 2023 to US$11 billion in 2028, driven by the increase in the number of PFA procedures.
The growing adoption indicates that PFA has the potential to become the preferred method for treating AFib over the existing treatments, such as thermal catheter ablation and surgical ablation procedures.
Initial clinical trials indicate PFA results in better patient outcomes
With this new technology, patients will experience an improved quality of life with a significantly lower risk of complications and post-procedural discomfort.
This finds evidence in some of the studies performed by the industry. In January 2024, the European Heart Rhythm Association published a study comparing the performance of Boston Scientific’s Farapulse PFA system against thermal ablation systems in 1,572 patients across Europe. The study showed that 85% of patients who underwent PFA experienced overall freedom from AFib after one year, compared to 77% of patients who underwent thermal ablation procedures.
Reduced time of post-procedure care is PFA’s major advantage
With a duration of about 2 hours, the PFA procedure is shorter than thermal ablation, which takes 3-4 hours. More importantly, PFA requires a few hours of hospitalization post the procedure, while thermal ablation is typically associated with one day of hospitalization after the procedure.
Shorter hospital stays improve patient experience by minimizing stress and discomfort from longer hospitalization hours. They also enable faster scheduling, as hospitals can perform more procedures and minimize scheduling delays.
As PFA does not require in-patient admissions, PFA procedures will not be disrupted by hospital bed shortages. This is a considerable advantage, as many developed countries such as the USA and the UK lack adequate hospital bed capacity. As of 2021, there were 2.8 hospital beds per 1,000 population in the USA and 2.4 in the UK, below the WHO’s recommendation of 3.4 beds per 1,000 population.
Moreover, reducing the length of hospital stays yields significant cost savings for patients as well as the payers. Reducing a hospital stay by a day or several hours translates to savings that cannot be ignored. For instance, in the USA, the average cost of per-hour hospital observation is US$600 in 2024, as per the healthcare pricing transparency platform Turquoise Health. The average cost of per-day hospitalization was US$2,883 in 2021, as per a study by the Kaiser Family Foundation (Medicare patients are eligible for $1,632 reimbursement). In the UK, the average cost of per-hour hospital observation is US$100, and the cost of per-day hospitalization is US$442 as of 2022, according to the National Health Service.
Short learning curve and procedure time facilitate performing more procedures
A short learning curve equips more cardiologists and trainees with the skills required to perform and support the procedure faster. Cardiologists typically get comfortable with PFA procedures after 5-10 cases, which allows to expand the pool of specialists performing this treatment relatively quickly and easily. This, in turn, can significantly improve PFA accessibility.
As the shortage of physicians continues to worsen globally, particularly in the USA, which represented 50% of the ablation market in 2023, PFA can play a crucial role in facilitating an increase in the number of procedures performed at a hospital within the same timeframe. With an expected shortage of 120,000 cardiologists in the USA by 2030, according to a 2021 report by the Association of American Medical Colleges, performing quicker procedures can help to partially offset the lack of specialists. Since PFA takes 30-50% less time than conventional ablation methods, it has the potential to significantly increase the number of procedures performed.
MedTech companies grow their ablation market share by offering PFA devices
With increased health screening efforts that detect more patients with arrhythmias, the number of cardiac ablation procedures performed globally doubled between 2013 and 2023 to reach about 650,000 procedures in 2023.
Boston Scientific expects the global AFib ablation market to more than double to US$11 billion during 2023-2028, with PFA predicted to grow to more than 80% of procedures (from under 5% in 2023). PFA technology is expected to be adopted quickly. As seen in Europe, PFA devices were launched in 2021, and already about 12% of the ablation procedures in the region in 2023 were done using PFA technology.
J&J, Medtronic, and Boston Scientific take the lead in the PFA field
Eyeing the potential of this emerging market, MedTech giants such as Johnson&Johnson (J&J), Medtronic, and Boston Scientific (accounting for 55%, 10%, and 5% share of the global thermal ablation market in 2023, respectively) have entered the market with their newly developed PFA devices. Being early entrants, these companies have the potential to expand their market shares in the cardiac ablation market by grabbing shares from thermal ablation procedures.
Boston Scientific was the first company to commercialize PFA devices with the launch of the Farapulse PFA system in Europe in January 2021. Boston Scientific enjoyed a two-year monopoly in the European market until Medtronic launched an integrated mapping and PFA system called Affera in March 2023. Later, the company launched another PFA system, PulseSelect, in December 2023. In February 2024, J&J’s Varipulse PFA system also received approval in Europe.
In the USA, Medtronic was the first company to receive FDA approval for its PFA system PulseSelect in December 2023, followed by Boston Scientific in January 2024. Medtronic also received FDA approval for Affera in March 2024.
J&J is the only company with a presence in Asia, as the company received approval for its PFA system in Japan in January 2024. Abbott is currently conducting clinical trials for its PFA system Volt in Australia and expects to start clinical trials in the USA this year.
The companies work to enhance and improve their systems. For instance, Medtronic’s integrated mapping and PFA system Affera offers enhanced procedure performance supported by real-time mapping. The integrated system includes an ablation catheter Sphere-9 and mapping software to facilitate real-time mapping. Sphere-9 catheter can perform high-density mapping and ablation simultaneously to allow cardiologists to deliver wide-area focal ablation lesions quickly. Affera can also work with the PulseSelect PFA system to provide real-time mapping. Similarly, J&J has a 3D mapping system called Carto 3 (in the market since 2009), which integrates well with its PFA system and generates real-time 3D mapping that aids in better cell targeting. Boston Scientific has not developed an exclusive mapping system for its PFA system, however, the company claims that any catheter mapping system will work well with Farapulse.
Comparing the PFA systems’ performance in the clinical trials, all systems, including Boston Scientific’s Farapulse, Medtronic’s PulseSelect, Medtronic’s Affera, and J&J’s Varipulse proved to be effective in over 70% of patients in terms of freedom from arrhythmia recurrence in one year.
Currently, PFA devices are only available in the USA, Europe, and Japan, with Boston Scientific dominating in Europe. Boston Scientific has witnessed high adoption rates in Europe so far, and the company has been able to serve 40,000 patients in three years since its entry into the European market in 2021. The company expects an overall organic sales growth of 8-10% during 2024-2026, driven by its PFA devices. Medtronic and J&J have just launched their PFA systems in the USA and Europe, and how these companies perform has yet to be seen. Analysts from BTIG financial services firm predict that Medtronic’s PulseSelect will secure 9% and Boston Scientific’s Farapulse will secure 14% of the cardiac ablation market (which comprises PFA and two other forms of thermal ablation procedures – radiofrequency and cryoablation) in the USA by 2025.
With competent technologies, the market is expected to witness stiff competition from these companies. Analysts from BTIG financial services firm predict that by 2027, PFA will grab 48% of the US cardiac ablation market, while the radiofrequency ablation market will have a 42% share and cryoablation a 10% share. The expected PFA’s 48% market share is likely to be split amongst the leading PFA systems – Boston Scientific’s Farapulse, J&J’s Varipulse, Medtronic’s PulseSelect, and Medtronic’s Affera, at 16%, 13%, 10%,7%, respectively, followed by others with 2% share.
While these companies have already entered the PFA space, Abbott’s wait-and-see approach to PFA may backfire on its performance in the EP market. The company aims to commercialize its PFA system Volt in the USA by 2027 or 2028. However, PFA’s fast adoption threatens Abbott’s US$1.9 billion EP business and its 15% global thermal ablation market share (as of 2023). Growing PFA adoption could also threaten Abbott’s diagnostic catheter and mapping systems, as healthcare providers using PFA systems would prefer buying mapping systems linked to PFA.
New entrants to drive innovation and further improve PFA technology
Apart from the large players, there are a few smaller players, such as Canada-based Kardium, US-based Adagio Medical, and US-based Pulse Biosciences, that are developing PFA systems. These companies are investing in improving the PFA using nanotechnology and supportive systems such as 3D mapping systems. For instance, Pulse Biosciences developed Nanosecond PFA (nsPFA) technology that uses superfast nanosecond pulses of electrical energy that can regulate cell death, which spares adjacent noncellular tissue. The company expects FDA approval for this system in 2024.
EOS Perspective
Over the years, MedTech companies have been actively pursuing the development of minimally invasive procedures that have shorter recovery periods, offer improved patient outcomes and reduced post-procedure discomfort. As the limitations of the existing ablation methods became apparent, PFA poses a vast growth potential, as it is a safer, more convenient, and more effective alternative.
On the other hand, the pulsed-field waveform is significantly more complex than the energy modalities that preceded it, with numerous variables determining the dose targeted at the tissues and the quality of the resulting lesion. While a variety of PFA systems have demonstrated effective ablation procedures, these systems have yet to advance in overcoming all limitations of targeting the tissue of interest and rare but potentially serious complications.
In the coming years, we can expect companies to develop multiple catheter configurations that allow cardiologists to configure the energy delivery to achieve the desired energy dose and lesions. This includes the development of multi-configurable ablation catheters that can shift shapes to create circular, linear, or focal ablation lesions without performing catheter exchanges.
As the technology advances, we can expect PFA to dominate the AFib ablation market and democratize AFib ablation procedures by improving accessibility to all eligible patients.