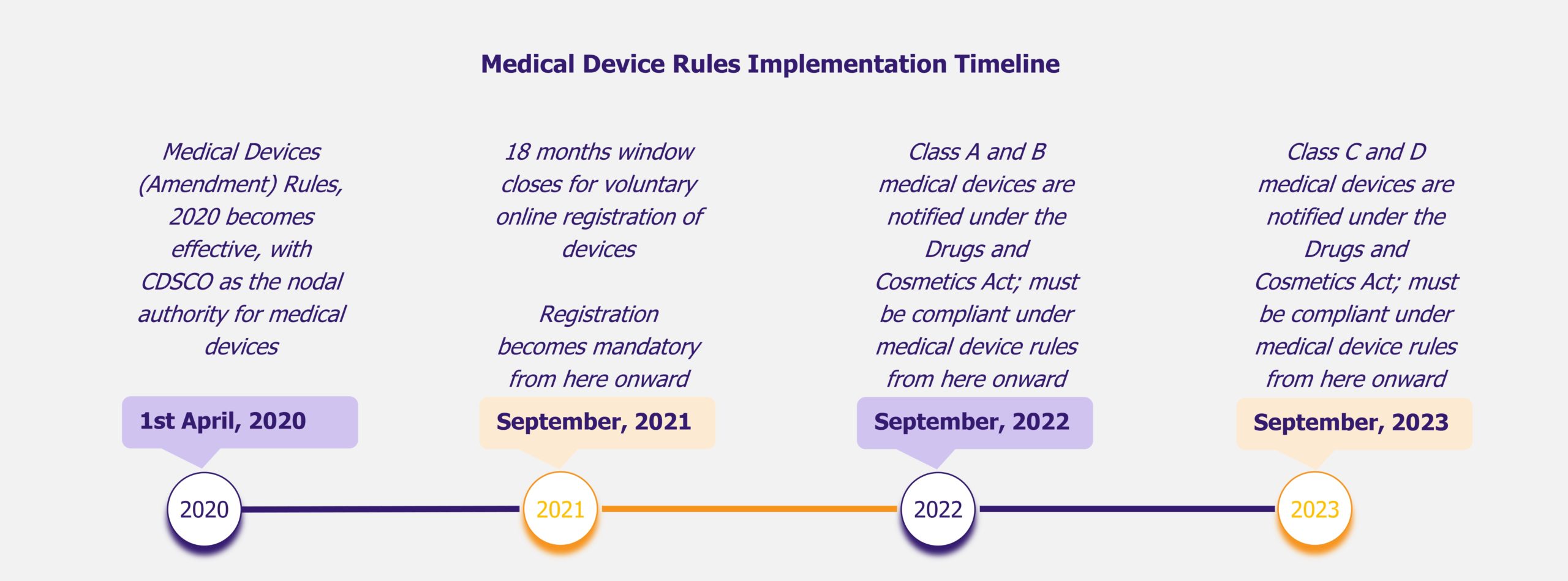
Although the COVID-19 pandemic seems to be done with its rampage, many people still opt to access all kinds of services, including healthcare, from the comfort of their homes. As this trend is expected to continue, the global digital therapeutics market, with its projected growth at a 20% CAGR from 2022 to 2035, is one important sector healthcare firms should focus on right now.
Digital therapeutics (DTx) are digital health interventions or software applications that are clinically validated and designed to treat or manage medical conditions. They can be used alone or in conjunction with traditional medical treatments.
The Digital Therapeutics Alliance categorizes DTx products into three types: disease treatment, disease management, and health improvement.
Examples of DTx include a solution to manage chronic musculoskeletal pain developed by Kaia Health, a biotechnology company in New York. This motion analysis tool assesses and guides patients’ progress during physical therapy and tailors treatment to individual requirements.
Similarly, Clickotine from Click Therapeutics, a company also based in New York, uses AI to help people with nicotine addiction. This solution offers a personalized plan fully integrated with eight weeks of nicotine replacement therapy, including options such as gum, patches, or lozenges. It tracks critical aspects such as daily cigarette counts, craving triggers, craving times, etc. A trial study conducted by the company in 2016 claimed that 45% of Clickotine users were able to quit smoking.
Adoption of DTx is taking off amid increased investments
The commercial development of DTx started around 2015 and, since then, has grown into a global market of considerable size. The total value of global DTx start-ups was estimated at a whopping US$31 billion in 2022, according to a 2022 report published by Dealroom, an Amsterdam-based firm offering data and insights about start-ups and tech ecosystems, in partnership with MTIP (a Swiss-based private equity firm), Inkef (an Amsterdam-based early-stage venture investment firm), and Speedinvest (an Austrian early-stage investor).
The number of people using DTx solutions is expected to increase over the next few years, according to a 2022 report by Juniper Research, a UK-based research firm. The study found that there were 7 million DTx users in the USA in 2020, a number expected to rise to around 40 million in 2026.
This increase can be attributed to the fact that DTx solutions are highly accessible and distributable due to an increase in the use of smartphones. A 2021 report published by Pew Research Center, a US-based think tank, found that 87% of Americans owned a smartphone in 2021, compared to 35% in 2011. With this, more people will be able to access medical care without having to spend more on hospital visits.
DTx applications have also been attracting numerous investors owing to the applications’ cost-effectiveness, ease of distribution, and better accessibility. According to the same 2022 report published by Dealroom, global venture capital funding in DTx witnessed a fourfold increase in 2022 compared to 2017.
All these studies reveal that, despite certain challenges, the DTx applications hold the promise of developing into a practical and affordable means of treating illnesses and conditions that impact large numbers of people.
Regulatory pitfalls present a major roadblock to DTx adoption
One main challenge DTx companies face is the regulatory environment. All DTx products must comply with the regulations of regional agencies such as the FDA, HIPAA, HITECH, etc.
Many US firms initially faced regulatory obstacles and payer resistance around product reimbursement. Before 2017, the US FDA classified DTx solutions as a SaMD (Software as a Medical Device) and, therefore, made them subject to risk assessment (low, medium, or high). Due to this, DTx solutions needed premarket approval and rigorous clinical trial results to get approval.
This has improved with the introduction of the Digital Health Innovation Action Plan by the FDA in 2017. According to the new plan, the FDA will first consider the company producing the solution. If the producer has demonstrated quality and excellence, it can market lower-risk devices with a streamlined premarket review. Post-market surveillance and data collection are also done to evaluate product efficiency.
Similarly, in the EU, DTx is controlled by national competent authorities and governed by the European Regulation on Medical Devices 2017/745 (MDR). However, no specific framework indicates the evidence required for assessing the performance or quality of DTx solutions or their production standards. This means that the member states may interpret the dossier requirements differently, leading to a fractured regulatory environment.
The COVID-19 pandemic has provided companies with some regulatory flexibility, leading to an increase in venture capital funding. In 2020, the federal government in the USA issued a new rule allowing healthcare practitioners to treat patients across state lines, including the use of digital medicine. This can increase access to healthcare, especially in rural areas, and physicians will be able to offer timely care to their patients traveling in a different state.
The FDA has also loosened regulations during COVID-19, particularly for mental health products, with the Digital Health Innovation Action Plan. This was to ensure that patients received timely care even from their homes while reducing the burden on hospitals. It waived certain regulatory obligations, such as the need to file a 510(k) premarket notification during the COVID-19 pandemic. The 510(k) is a submission indicating that a new medical device is similar to something already approved by the FDA (a predicate device) to ensure safety and efficiency. However, finding suitable comparables can be highly challenging in the case of DTx, which is dynamically evolving. This can result in misunderstandings or overlooking of critical aspects of these solutions, leading to uncertainty and delays in the approval process. The waiver of this regulation offers DTx companies some relief in the future.

Digital Therapeutics – The Future of Healthcare by EOS Intelligence
Patient health literacy is a hurdle in the adoption of DTx solutions
A survey by the National Assessment of Adult Literacy (NAAL) in 2003 has shown that only 12% of Americans possess proficient health literacy skills, making them able to find and understand information related to their health. This lack of awareness among patients can also impede the ease of applying DTx products.
Patient experience is also crucial for the acceleration of DTx adoption. Older patients unfamiliar with using technological gadgets can find it difficult to adopt DTx solutions. However, a 2022 AMA survey has shown that 90% of people over the age of 50 in the USA recognize some benefit from digital health tools.
Similarly, a survey conducted by the Pew Research Center in 2021 indicated an increase in the use of smartphones and the internet among older people in the USA, driven by the pandemic. Older adults are using technological applications for activities such as entertainment, banking, shopping, etc., even after the pandemic, a 2021 survey by AARP Research, a US-based NPO, shows. This indicates that there is scope for an increase in adoption.
Many companies are now trying to increase patient involvement by using gamification, aiming at patient groups for whom DTx use is likely to be more challenging (e.g., older population, children). DTx developers include game-like elements or mechanics into a DTx solution, such as tasks, rewards, badges, points, and leaderboards. An example is US-based Akili Interactive’s EndeavorRx, a prescription DTx aimed at enhancing attention function in children with ADHD aged 8 to 12. It uses an interactive mobile video game to assist children in improving their attention skills and adjusting to their performance levels. The game’s sensory stimuli and motor challenges also help kids multitask and tune out distractions.
Payer reluctance affects many DTx products
Although the number of DTX products on the market increases, payers’ reluctance to cover their costs to the patient can also slow down adoption. The coverage of DTx solutions is limited, even when they are FDA-approved. Only 25% of payers are currently willing to cover prescription DTx solutions, according to a 2022 survey by MMIT, a Pennsylvania-based market data provider, which involved 16 payers.
Akili Interactive’s EndeavorRx is one such solution facing insurance coverage issues. Elevance Health (previously Anthem) denied coverage for EndeavorRx, deeming it medically unnecessary, while Aetna, another insurance provider, considers it experimental and investigational.
A study released by Health Affairs, a health policy research journal, in November 2023 has shown that only two of the twenty FDA-approved prescription DTx solutions on the market have undergone rigorous evidence-based evaluation. This means that no authoritative results indicating the benefits of these solutions for various population demographics are available, making many payers skeptical of their medical claims.
DTx offers solutions for managing multiple conditions
Over the past few years, several prominent players have emerged in the DTx landscape. Around 59% of the DTx market is concentrated in the North American region and 28% in Europe.
Top players, such as Akili Interactive and Big Health, both US-based firms, focus on offering products for managing mental health illnesses, mostly management of anxiety, depression, and stress, according to a report published in 2023 (based on data until September 2022) by Roots Analysis, an India-based pharma/biotech market research firm. With about 970 million people suffering from mental health conditions globally (according to the WHO), the potential user pool is enormous, offering growth opportunities for DTx solutions developed to address mental illnesses and, over time, driving the growth of the DTx market as a whole.
Many top companies also focus on solutions offering pain management and treatment for chronic conditions such as diabetes, obstructive pulmonary disease, and musculoskeletal disorders. An example is US-based Omada’s pain management solution, Omada MSK. This application guides patients through various customized exercises and records their movements, which are then assessed by a licensed physical therapist (PT), who can make recommendations for improvement. It also has a tool that utilizes computer vision technology to help PTs virtually assess a patient’s movement and range of motion, allowing them to make necessary changes in the therapy.
Similarly, several DTx solutions on the market now focus specifically on diabetes, which affects around 537 million adults globally. Some top companies focus on the previously unmet needs of conventional methods, such as weight management or preventing prediabetes, to help with overall diabetes treatment. US-based Omada’s solution, Omada Prediabetes, comes with a weight scale pre-connected to the app, and the weight is added to the app as soon as the patient steps on the scale. A dedicated health coach assesses the patient’s weight, creates a customized plan, and monitors the patient’s progress. In other similar DTx solutions for diabetes, an app can also give insulin dose recommendations based on the patient’s blood glucose levels.
DTx can serve in a range of other conditions, including major depressive disorder, autism spectrum disorder, and multiple sclerosis, to name a few.
The DTx landscape is rife with development
The DTx business landscape has recently seen many developments, from acquisitions to product launches. One of them was Big Health’s acquisition of Limbix, a California-based DTx firm, in July 2023 to bolster its portfolio, including SparkRx, a treatment for adolescents dealing with depression and anxiety. Similarly, in June 2023, Kaia Health launched Angela, a HIPAA-compliant, AI-powered voice-based digital care assistant, to serve as a companion and guide, enhancing the physical therapy experience for patients.
In another development, BehaVR, a DTx company headquartered in Kentucky, and Fern Health, a digital chronic pain management program, merged their companies in November 2023 to create a novel pain management DTx solution that addresses both pain and fear caused by chronic diseases. With this merger, they launched RealizedCare, an app designed to offer a comprehensive solution that collaborates with health plans, employers, and value-based providers to treat a range of behavioral and mental health conditions. This solution provides clinicians with immersive programs specifically designed for in-clinic use. It is initially focusing on chronic pain.
Bankruptcy of Pear and lessons for the industry
However, the most shocking development in the DTx market was the bankruptcy of Pear Therapeutics in 2023. The remains of this once-prominent company were purchased by four other companies for a total of US$6.05 million at an auction. Pear was a big name in the industry since its inception in 2013. It introduced numerous products such as reSET, reSET-O, and Somryst for treating substance use disorder, opioid use disorder, and chronic insomnia, respectively. It was also the first company to receive FDA approval for a mobile app aimed at treating substance use disorders.
Though the company announced layoffs of nearly 20% of its workforce in November 2022, its management expressed optimism about the company’s growth and reduced operating expenses in the third quarter. But in April 2023, the company filed for bankruptcy.
The demise of Pear has opened the eyes of industry experts to the challenges faced by DTx players. Certain issues were unique to Pear itself, such as the comparatively higher prices of its products and the focus on treating challenging conditions such as substance use disorders. However, the bankruptcy of Pear also brings attention to the obstacles that can be faced by any other DTx company. One crucial roadblock is that physicians and payers still approach these products with caution. Additionally, achieving profitability for DTx might be challenging for all types of players, particularly for small start-ups lacking substantial market influence. The bankruptcy of Pear and the challenges it faced can be used by budding DTx companies as a road map as they navigate this complex sector.
EOS Perspective
DTx is all set to revolutionize the medical industry, with a 2020 McKinsey report suggesting it could potentially alleviate the global disease burden by up to 10% by 2040. Given the impact of emerging treatments on stakeholders, pharmaceutical and healthcare companies should consider expanding their portfolio to include DTx solutions.
With telehealth companies seeing good growth in the pandemic and post-pandemic years, an increase in investment can be expected as they are uniquely placed to support prescription DTx. With the growth of the digital health industry, prominent telehealth providers may also choose to acquire DTx businesses or create their own in-house DTx solutions.
Read our related Perspective: COVID-19 Outbreak Boosts the Use of Telehealth Services
An increase in industry M&A activities can be expected in the next few years, with growing incidences of chronic illnesses, improved technology penetration across all age groups, and a maturing market. Big names such as Bayer, Novartis, and Sanofi are also entering into partnerships with DTx companies, indicating a bright future for the sector.
Mental health and behavioral therapy are great fields to branch out for companies starting in the DTx landscape, especially in this post-pandemic era. Demand for such services is likely to be sustained, considering the National Institute of Mental Health Disorders estimates that one in four adults in the USA suffers from a diagnosable mental illness, with many suffering from multiple conditions.
Similarly, diseases such as diabetes, cancer, heart, and respiratory ailments are on the rise. Healthcare companies can effectively address these medical areas through the use of DTx applications, providing personalized care for patients. This approach has the potential to manage not only chronic conditions such as diabetes but also terminal illnesses such as cancer.
Many DTx players will likely focus on areas with unmet needs, including pediatrics and metabolic disorders. With seven DTx-based diabetic management solutions already receiving 510(k) clearance as of December 2022, it can be expected that more products addressing the treatment gaps might flood the market.
The DTx industry is gradually maturing and has been receiving significant investments in recent years (US$8 billion in 2022). While experts view it as a profitable market, hesitation remains, particularly following the bankruptcy of Pear Therapeutics.
Nevertheless, due to the COVID-19 pandemic and subsequent lockdown measures, technology adoption among older adults has increased significantly. Hence, strategic investments in DTx by pharmaceutical and healthcare companies, taking into account market conditions, can expect to establish a stronger presence in this industry in the future.