The market reaction to the US Inflation Reduction Act of 2022 is mostly mixed. It is expected to change the pharma industry dynamics in terms of the competitive positioning and product pricing of those companies projected to be negatively impacted by the IRA. The answer to whether the IRA will be able to curb rising healthcare costs in the USA lies in the legislation’s on-the-ground application.
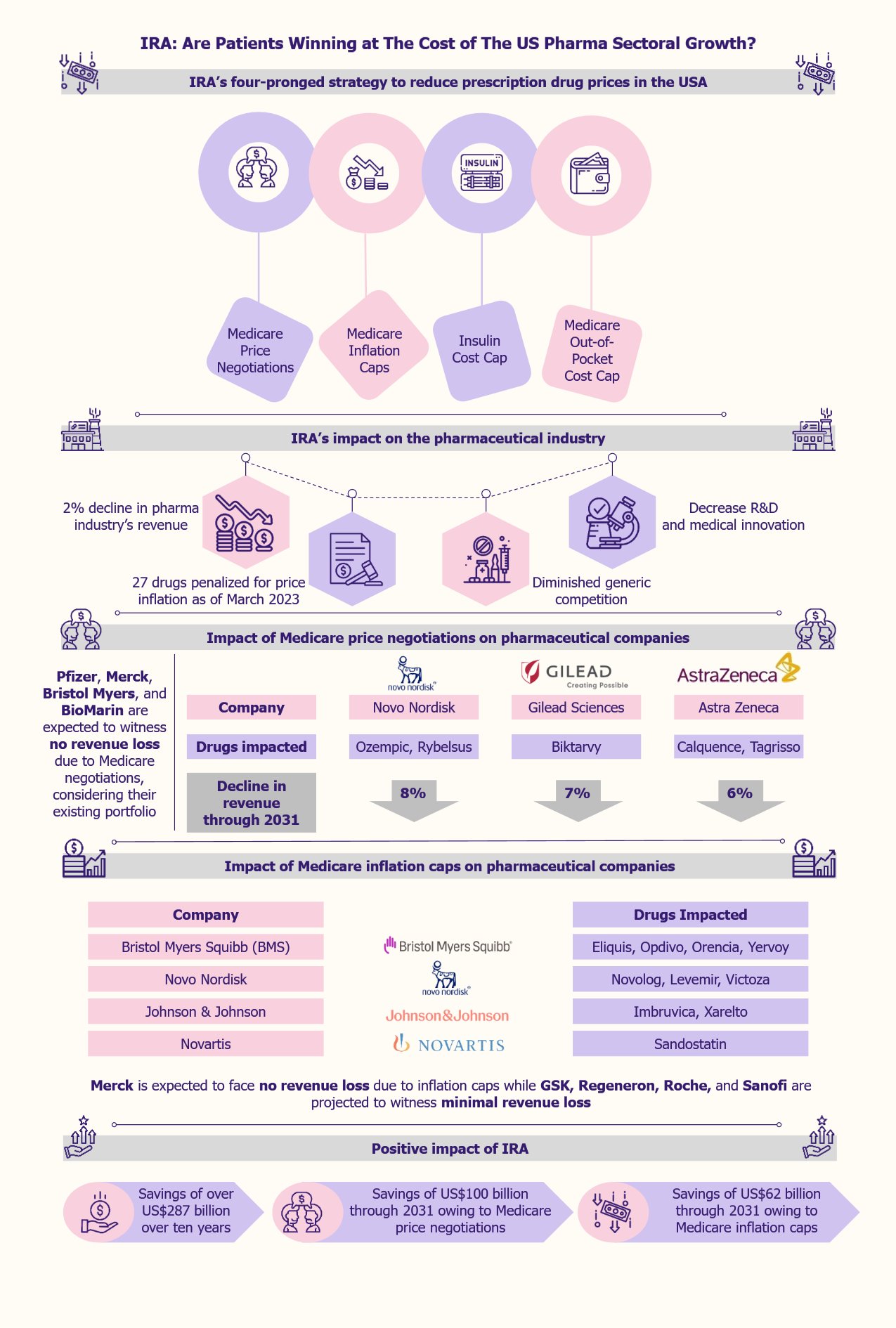
IRA to decrease prescription drug prices via a four-pronged strategy
Prices of prescription drugs in the USA are 2.78 times higher than in 33 other countries analyzed in a 2024 report published by RAND, a public policy think tank.
In pursuit of reducing healthcare costs in the USA, the Biden government passed the Inflation Reduction Act (IRA) in August 2022. One of the major goals of the act includes the reduction of prices of prescription drugs.
This is expected to be achieved through a four-pronged strategy, the mainstay of which involves the US federal government negotiating the prices of some high-priced prescription drugs covered under Medicare.
The second prong includes pharmaceutical firms paying a rebate to Medicare if they raise the price of prescription medicines covered under Medicare by a rate that is higher than the inflation rate.
The monthly cost of insulin for Medicare patients is capped at US$35, as the third prong.
The fourth prong aims to reduce prescription drug prices by capping the out-of-pocket costs of Medicare Part D patients at US$4,000 in 2024 and US$2,000 in 2025.
IRA Are Patients Winning at the Cost of the US Pharma Sectoral Growth by EOS Intelligence
Pharma companies to suffer more due to IRA compared to projected government savings
Under the IRA, large pharmaceutical companies, defined as those with over US$1 billion in net profits, are required to pay a minimum of 15% annual taxes, a financial burden on these companies. Analysts predict that the annual revenue from corporate taxes could be to the tune of US$222 billion. Furthermore, the IRA is expected to save over US$287 billion for ten years from the roll-out, as per the estimates of the Congressional Budget Office (CBO).
Apart from the increased financial burden on some companies, experts foresee potential adverse impact on several pharmaceutical companies based in the USA to a considerable extent.
The pharma companies witnessing the least to no impact are the ones with their primary operations based outside the USA, biologics or large molecule drug producers, and the ones that do not receive government funding for R&D. This is because of the differing timelines under IRA for negotiating the prices of biologics and small molecules. Biologics’ timeline is 11 years after FDA approval, while small molecule drugs are eligible after 7 years. Therefore, Medicare negotiations will begin four years earlier for a small molecule drug that has received approval at the same time as a large molecule biologic drug.
Apart from these adverse effects, such as differential treatment of small molecule drugs compared to biologics under Medicare price negotiation timelines, there are some other negative impacts on the overall US pharma industry, such as diminishing competition among generic drug producers, decreased discovery of new treatments, and new uses of existing drugs.
IRA to affect the revenues of top pharma companies surely but variably
There are differing viewpoints regarding the impact of IRA on pharmaceutical companies’ revenue. One group of experts suggests that Medicare prescription drug negotiations under the IRA will depend on the expiration of the drug’s patent. Other experts expressed their opinion that irrespective of when a drug loses exclusivity, a significant threat to drug revenues comes from the competition entering the market and not from lower negotiated drug prices.
The first group of experts states that lower negotiated prices in 2026 are expected to have a lower impact on medicines projected to witness revenue loss owing to patent expiry around the same time. One such example of a drug losing its exclusivity in the USA in 2025 is Stelara by Janssen Biotech approved for treating psoriasis.
In contrast, pharma companies producing medicines that are expected to witness competition from their generic counterparts after 2026 are projected to lose revenue owing to lower negotiated prices even before the drugs lose exclusivity. However, some companies’ revenue will be affected more than others.
Medicare price negotiations to hit revenues of some drugmakers drastically
The pharma industry’s revenue is expected to decrease by 2% due to the new measures brought about by the IRA, as per a 2022 report by Morningstar, a US financial services firm. Among the companies that will be highly affected are Novo Nordisk, Gilead, Bristol Myers Squibb, AbbVie, and AstraZeneca. In contrast, others, such as Pfizer, Merck, Roche, and Novartis, will not be as much impacted by Medicare price negotiations.
Some 15% of global branded drug sales come from Medicare in the USA, as per Morningstar estimates. Therefore, the impact of the IRA on pharmaceutical companies depends on their reliance on Medicare sales, price adjustments, high-cost specialized drugs, and extended patent protection.
Medicare prescription drug negotiations are projected to impact pharma companies the most among all IRA measures, although this impact might not be uniform across the players. On the other hand, Medicare negotiations are projected to save the government approximately US$100 billion through 2031. The pharma companies facing the highest revenue losses include Novo Nordisk, Gilead, and AstraZeneca.
When the Medicare price negotiation measures start to roll out in 2026, two drugs of Novo Nordisk, namely, Ozempic and Rybelsus, that are approved to treat type 2 diabetes, are expected to witness an 8% decline in their projected revenue through 2031, as per Morningstar. Gilead’s Biktarvy, which treats HIV-1 infections, is expected to be subject to price negotiation in 2027 and thereby face a projected revenue loss of 7% through 2031. On similar lines, Calquence (to treat mantle cell lymphoma) and Tagrisso (to treat non-small cell lung cancer) drugs of AstraZeneca are expected to lose 6% revenues through 2031 owing to Medicare price negotiations.
In contrast, considering the existing portfolios, Pfizer, Merck, Bristol Myers, and BioMarin are expected to witness no revenue loss due to Medicare negotiations.
Medicare inflation caps to impact major pharma companies negatively
Another important IRA measure is Medicare inflation caps. This measure involves drug producers paying penalties for increasing drug prices beyond the inflation rate. It is expected to result in US$62 billion in government savings through 2031.
Around March 2023, the US federal government, along with the Centers for Medicare & Medicaid Services (CMS), released a list of 27 drugs whose prices were increased by their manufacturers at a higher rate than the inflation rate. This list included AbbVie’s Humira (to treat Crohn’s Disease) and Astellas Pharma’s and Seagen’s Padcev (to treat urothelial cancer). Gilead Sciences, Johnson & Johnson, and Pfizer are among other impacted companies by Medicare inflation caps. Pfizer had the most drugs on the list, with a total of five.
Bristol Myers Squibb is one of the pharma companies that is expected to be highly impacted by Medicare inflation caps. The company’s drugs, such as Eliquis (to treat or prevent blood clots), Opdivo (to treat melanoma), Orencia (to treat rheumatoid arthritis), and Yervoy (to treat various cancer types) are among the medicines that are expected to face revenue loss owing to inflation caps. Other drugs on the list include Novo Nordisk’s drugs such as Novolog and Levemir (both for type 1 diabetes) and Victoza (for type 2 diabetes), Johnson & Johnson’s drugs such as Imbruvica (to treat certain cancers) and Xarelto (to treat or prevent blood clots), along with Novartis’s Sandostatin (for severe diarrhea and flushing related to metastatic carcinoid tumors).
In contrast, Merck is not expected to face any revenue loss due to inflation caps, while GSK, Regeneron, Roche, and Sanofi are projected to witness minimal revenue loss as these companies have not raised the prices of their drugs beyond the inflation rate.
IRA to potentially reduce competition from generics
According to the IRA, following the price negotiations of some of the branded drugs, manufacturers of the generic versions of such drugs will have less scope to charge a reduced price for those drugs. This would disincentivize the generic drug producers to manufacture generic versions of the already low-priced branded drugs.
EOS Perspective
The IRA represents a substantial change in the US legislation that strives to make healthcare more affordable to Americans through increased access to more reasonably priced prescription medicines.
However, IRA can be expected to affect small-molecule drugmakers more negatively than biologics. Moreover, some pharmaceutical companies are projected to feel the pinch more than others in terms of revenue losses.
Companies such as Merck, Bristol Myers Squibb, and the pharmaceutical association PhRMA have filed lawsuits against some provisions of the IRA, stating that they are unconstitutional. Bristol Myers Squibb and J&J are planning to appeal after the US court dismissed the IRA lawsuits. These pharmaceutical companies are trying to find ways to circumvent the negative impact of the legislation.
IRA is also expected to negatively impact R&D and medical innovation. This is evident from the fact that biopharma companies have reduced their R&D efforts in the neuroscience space, especially since a lot of development work in this space involves small-molecule drugs. Moreover, as IRA exempts only one orphan drug from price negotiation, investments in R&D for orphan drugs are likely to get deprioritized. Many pharmaceutical companies are reconsidering their R&D planning and investment strategies to counter the effect of IRA.
IRA is clearly not a win-win strategy for all stakeholders. Pharmaceutical companies are mostly at the losing end, while patients could be winners. Considering all the positives and negatives of IRA, only time will tell the actual impact of the legislation on the overall pharmaceutical industry.